Position Frequency Analysis (PFA)
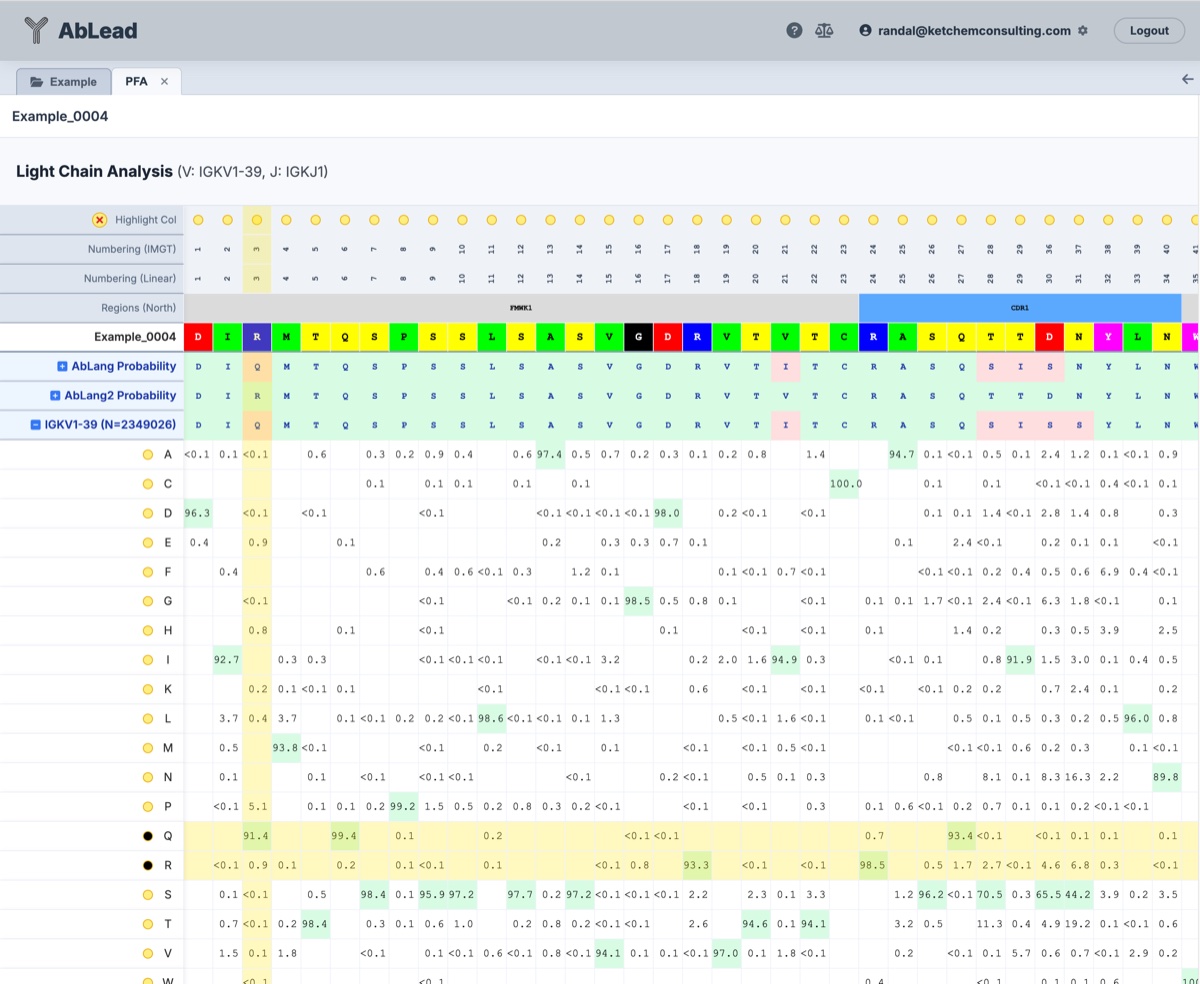
The PFA view provides a statistical and LLM-based breakdown of amino acid distributions at each position.
This tool is useful for identifying specific likelihoods and probabilities for each position in the antibody sequences. It is particularly useful for selecting mutations in antibody engineering and developability optimization. Note that human germlines are searched regardless of parent species.
PFA is a great example to Be Inherently Lazy. Pulling together the individual LLM grids and statistics on the closest human germlines manually would take a long time and is prone to error. Let the tool handle it for you.
Accessing the Tool
Select exactly one antibody in the Project View. Go to the Analysis menu and select PFA. This will open the PFA workspace in a new tab.
Using the Tool
- Liabilities: A liabilities row above the sequence indicates residue liabilities that could require repair or closer consideration for engineering. An 'S' in a liability cell indicates a Severe stability violation.
- Coloring: Highest frequency or likelihood residues are indicated by red highlighting of the sequence.
- Germline: Evaluate the closest germline. Use the Germline tool for more detail.
- Grid Exposure: Open a grid to visualize the probabilities for all residues at each position. Note that human germlines are searched regardless of parent species.
- Engineering Mutation Designs: Click a residue to enter a mutation design used by the Engineering tab.
- Observations: Click a residue to enter an observation for that IMGT position. These are displayed in the Observations tab and on a clicked residue.
- Export Excel: Click to export an Excel version of the grids.
- Indicator Dots: Dots on a residue position indicate that there is an associated engineering design or observation at that position. Engineering mutations will appear as a small eggshell square while observations will appear as a small orange circle.
The AntPack training set of 58,788,431 heavy chain and 68,454,444 light chain human sequences was used to calculate the PFA values.
The AbLang, AbLang2, and IgBert grid values are generated by the models as residue likelihoods. A higher positive value indicates a higher likelihood of the residue at that position. The difference in the likelihood between the parental and highest likelihood residue indicates the severity of the difference, with large differences indicating a possibility that the parental position is not optimal and could be mutated to improve stability.
If no PFA data exists for the closest human germline, the tool uses the data of the closest human germline with data in the PFA data set. This is annotated in the chain title.
3D Structure Visualization & Residue Selection
The PFA workspace includes an interactive PDBe Mol* 3D structure viewer positioned below the grids, allowing real-time mapping of sequence changes to the three-dimensional antibody structure.
-
Interactive Resizer: Drag the horizontal divider bar directly above the 3D window up or down to adjust the vertical height of the viewer pane.
-
Cartoon Region Coloring: The ribbon structures of the light and heavy chains are colored by region to match the centralized color schemes:
- Variable Domain (Fv): Frameworks (dim gray/silver) and CDRs (blues, purples, cyans) are colored according to domain region standards.
- Constant Domains: Reconstructed constant domains (CL, CH1, hinge, CH2, CH3) are automatically rendered and colored by their specific sub-domain region colors.
-
Selected Residue Highlighting:
- Clicking column headers in the alignment grids (scheme position or linear number) highlights those columns in blue.
- The corresponding residues on the 3D structure are instantly highlighted in CPK element coloring (with position number labels), regardless of which exclusion method is active.
- Residue Style: Toggle the dropdown to change the rendering style of selected residues between Spacefill (default) and Ball-and-Stick.
References
- AbLang details are described in Olsen et al., Bioinformatics Advances (2022).
- AbLang2 details are described in Olsen et al., bioRxiv (2024).
- IgBert details are described in Kenlay et al., arXiv (2024).